Lo studio è stato condotto dall’Ospedale Pediatrico Bambino Gesù insieme all’Istituto Superiore di Sanità e ad altri centri internazionali. La patologia, della famiglia delle RASopatie, è dovuta a mutazioni del gene MAPK1 e all’iperattivazione della via di comunicazione intracellulare promossa da questa proteina

Roma, 29 luglio 2020 – Individuata una nuova sindrome del neurosviluppo causata dalla mutazione di un gene denominato MAPK1 e riscontrata a oggi in soli 7 bambini nel mondo. La patologia fa parte delle RASopatie, un gruppo di malattie rare di origine genetica caratterizzate da un quadro clinico che include bassa statura, dismorfismi facciali, deficit cognitivo variabile, un ampio spettro di difetti cardiaci, anomalie a carico dell’apparato scheletrico e anche una predisposizione all’insorgenza di neoplasie in età pediatrica.
La scoperta, effettuata da clinici e ricercatori dell’Ospedale Pediatrico Bambino Gesù, dell’Istituto Superiore di Sanità e di altri centri europei e statunitensi, è stata pubblicata sulla rivista scientifica American Journal of Human Genetics.
Le RASopatie
Le RASopatie sono un gruppo di patologie congenito-malformative causate dall’alterato funzionamento del meccanismo di comunicazione intracellulare mediato dalle proteine RAS, che controllano la crescita e la moltiplicazione delle cellule.
Queste proteine sono incaricate di trasmettere (trasdurre) le informazioni (per esempio gli stimoli di ormoni e fattori di crescita) dalla superficie della cellula al suo interno, attivando una serie di reazioni a cascata che costituiscono la via MAP-chinasi (MAPK).
L’eccessiva attivazione di queste proteine e della cascata di segnalazione RAS-MAPK, oltre a provocare i danni dello sviluppo, è tra le cause principali dell’insorgenza dei tumori, proprio per il ruolo chiave che queste proteine e l’intera via hanno nella regolazione di processi biologici quali la proliferazione e il differenziamento.
Lo studio
La nuova malattia identificata nello studio è causata in particolare dalle mutazioni di MAPK1 (noto anche come ERK2), che regola l’attività di numerose proteine presenti nella cellula. Le mutazioni di questo gene sono la causa della condizione osservata in 7 pazienti, seguiti dai diversi Centri internazionali, che condividevano un disordine del neurosviluppo associato a bassa statura, malformazioni cardiache e caratteristiche craniofacciali riconducibili alla sindrome di Noonan, una delle RASopatie più comuni.
La causa molecolare della malattia è stata identificata grazie all’uso delle nuove tecnologie di sequenziamento genomico nell’ambito di uno studio condotto all’interno del programma di ricerca “Vite Coraggiose” dell’Ospedale Pediatrico Bambino Gesù. Il programma è dedicato ai pazienti senza diagnosi ed è finanziato dalla Fondazione Bambino Gesù.
L’utilizzo di diversi approcci sperimentali basati su studi in vitro e in vivo, condotti grazie ai finanziamenti della Fondazione AIRC per la Ricerca sul Cancro, ha permesso di dimostrare come le mutazioni identificate siano alla base di una iperattivazione della proteina MAPK1 e dell’intera ‘cascata’ MAPK, cioè delle reazioni chimiche attivate dai segnali ricevuti dalle cellule. Lo studio ha anche permesso di formulare delle prime ipotesi sul perché queste specifiche mutazioni abbiano meno impatto nello sviluppo di tumori rispetto a quelle che colpiscono altre proteine della stessa cascata.
Il lavoro pubblicato sull’American Journal of Human Genetics è stato condotto da ricercatori e clinici dell’Ospedale Pediatrico Bambino Gesù, dell’Istituto Superiore di Sanità e di altri centri in Europa (Olanda, Spagna, Germania) e negli Stati Uniti (Indianapolis, New York, Missouri, Ohio, Maryland).
La strada percorsa
Sono passati 20 anni da quando è stato identificato il primo gene coinvolto nella più frequente tra le RASopatie, la sindrome di Noonan. Si trattava del gene PTPN11, alla cui scoperta contribuirono proprio le ricerche del dott. Marco Tartaglia, responsabile dell’area di ricerca Genetica e Malattie Rare dell’Ospedale Pediatrico Bambino Gesù.
Da allora, anche grazie all’importante contributo della rete italiana per le RASopatie e della rete europea NSEuroNet, da diversi anni finanziata dall’Unione Europea nell’ambito dei finanziamenti dedicati alle malattie rare (programmi E-Rare e EJP-RD), sono stati identificati numerosi altri geni responsabili della stessa sindrome di Noonan e delle altre RASopatie correlate.
“Il gene MAPK1 – spiega il dott. Tartaglia – era l’ultima proteina di questa importante via di comunicazione cellulare a non essere stata associata a una malattia genetica, quando mutata. Oggi si unisce alla famiglia dei geni implicati in una delle più frequenti famiglie di malattie genetiche che colpiscono lo sviluppo e la crescita dei bambini. Siamo contenti di poter aver potuto offrire un nuovo contributo nel campo delle malattie rare e nella comprensione dei meccanismi molecolari attraverso cui il malfunzionamento di questa proteina e della cascata RAS-MAPK altera i processi dello sviluppo e contribuisce all’oncogenesi”.
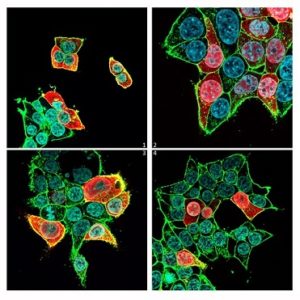
Fig. 1 – Le immagini raffigurano le cellule (contorno verde), i loro nuclei (in azzurro) e le proteine MAPK1 (colorate in rosso). Nella prima figura sono rappresentate le cellule sane, cioè con la proteina MAPK1 non mutata. Nelle altre, invece, si osserva la localizzazione delle proteine mutate nel nucleo, che lo fanno diventare rosso
 Salva come PDF
Salva come PDF